

BREAKING :: Leaked EU Regulatory Report Reveals Major Objections Preclude Marketing Authorisation for Pfizer BioNTech (BNT162b2) COVID-19 Vaccine November 2020
One major objection was identified that precludes a marketing authorisation three weeks before 'Authorisation' granted. "The characterisation of BNT162b2 DS is currently not found acceptable."

SUMMARY
As it conducted its analysis of the Pfizer-BioNTech Covid-19 vaccine in December, the European Medicines Agency (EMA) was the victim of a cyberattack. More than 40 megabytes of classified information from the agency’s review were published on the dark web, and several journalists—including from The BMJ—and academics worldwide were sent copies of the leaks. Here are the key points from those leaks.
Pfizer BioNTech (BNT162b2) COVID-19 Vaccine Safety & Efficacy Clinical Trial Data was performed only on Development Batches “Process 1” and is therefore INVALID
Drug product batches at the intended full commercial scale “Process 2” had NOT been manufactured at the time the clinical trials commenced and insufficient time was allowed for these DP Process 2 batches to be assessed in the clinical trials
Process 2 batches, with lower RNA integrity, were introduced in clinical trials but the date for the clinical Interim Analysis (IA) was changed, resulting in IA not including data from subjects dosed with “Process 2” material
European CHMP's assessors comprehensive scientific evaluation of the sponsors data revealed Major Objections Preclude Marketing Authorisation
The Development Material had RNA Integrity of 85% with In Vitro Expression of 67%
Post clinical trial material had RNA Integrity as low as 52% with In Vitro Expression as low as 49%
Leaked e-mails show the approval date was politically driven and set in stone with no wiggle room to address the process control and poor large scale RNA quality
Bridging studies (although suggested) were NOT done to ensure the scaled up manufacturing process produced the same safety and efficacy reported in clinical trials on the development batches
FDA/HC/EMA ‘agreed’ to accept 50% purity intact RNA so that not one region was exposed to SUB-OPTIMAL product being distributed
Ultimately, on 21 December, EMA authorised Pfizer-BioNTech’s vaccine
They knew that the development of a vaccine to express the original D614 form of the spike protein (original Wuhan-1 SARS-CoV-2 viruses) would have biological significance now that most circulating variants have the G614 form of the Spike protein.
Pfizer continued to tweak the manufacturing process resulting in a higher % of intact mRNA and a correlated higher number of deaths

Abstract
Drug product batches at the intended full commercial scale “Process 2” were manufactured too late to be included in the clinical trials as the interim analysis was cut short before they could reach the end point for inclusion in the IA…
Process 1 BNT162b2 product ≠ Process 2 BNT162b2 product and this raised serious concerns for the European CHMP's assessors comprehensive scientific evaluation of the sponsors data.
Committee for Medicinal Products for Human Use (CHMP), the European Medicines Agency's (EMA)* committee responsible for human medicines identified this as one Major Objection and raised it’s concerns with the applicant (Pfizer) …
After contact with the applicant it was confirmed that DP batches manufactured from early Process 2 batches, with lower RNA integrity, have been recently introduced in clinical trials. However, as the cut-off date for the clinical Interim Analysis (IA) was changed, the IA doesn’t include data from subjects dosed with Process 2 material, and the Company does not expect to have Process 2 included in the Final Analysis dataset.
Pfizer BioNTech (BNT162b2) COVID-19 Vaccine Phase I Pfizer BioNTech (BNT162b2) COVID-19 Vaccine Phase II/III clinical trials used to determine safety and efficacy were conducted on a different BNT162b2 product manufactured by “Process 1”. I/II Safety & Efficacy Clinical Trial Data is therefore completely invalid.
No bridging studies were done to ensure the scaled up manufacturing process produced the same product used in clinical trials.
… the Company does not expect to have Process 2 included in the Final Analysis dataset. Therefore, the proposed acceptance criteria of ≥50% intact RNA for RNA integrity is considered too wide compared to clinical batch data, 69-81%. The proposed release and shelf-life acceptance criteria for the DP should therefore be tightened based on the clinical data included in the dossier or clinically qualified by other means.
In-Development Batches of Pfizer BioNTech (BNT162b2) COVID-19 Vaccine
As seen below in a leaked document, the batch scale for drug substance construct BNT162b2 manufactured by “Process 1” were between 35 mL and 360 mL.
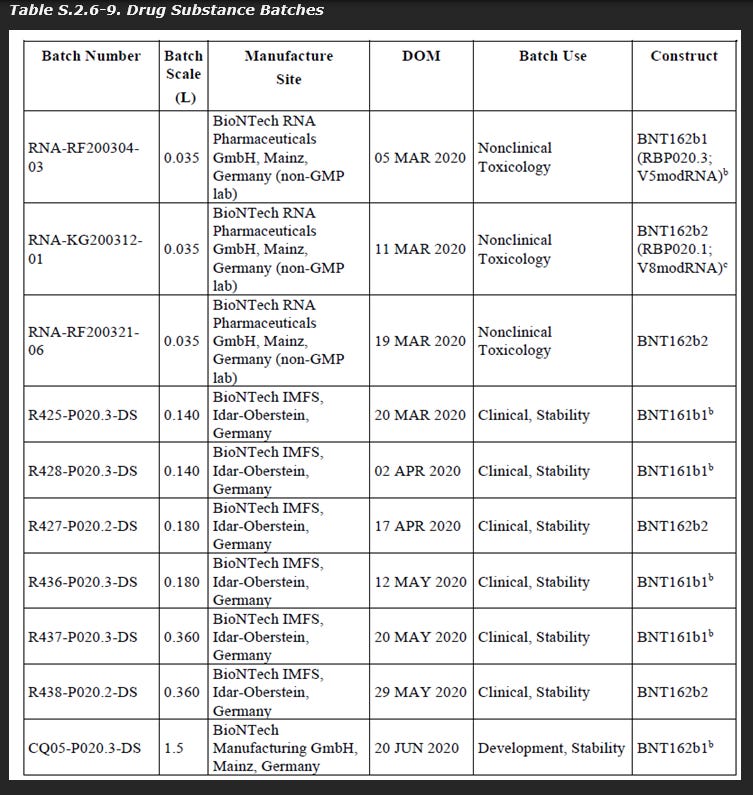
As seen below in another leaked document, the batch scale for drug substance construct BNT162b2 manufactured by “Process 2” used a starting volume of 37.6L.

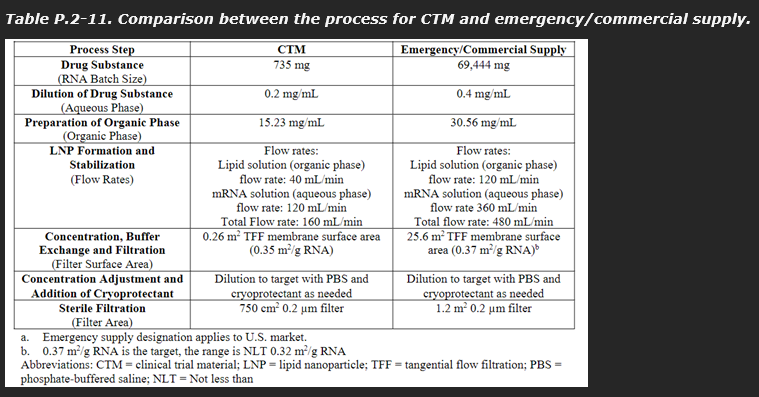
A comparison of the CTM and commercial supply variations other than batch size is summarised by the sponsor applicant in the table below.
The batches used as clinical trial material had RNA Integrity ranging from 55% - 85% with In Vitro Expression ranging from 50% - 71%.
The batches used as post clinical trial material had RNA Integrity ranging from 52% - 71% with In Vitro Expression ranging from 49% - 72%.
The Development Material had RNA Integrity of 85% with In Vitro Expression of 67%
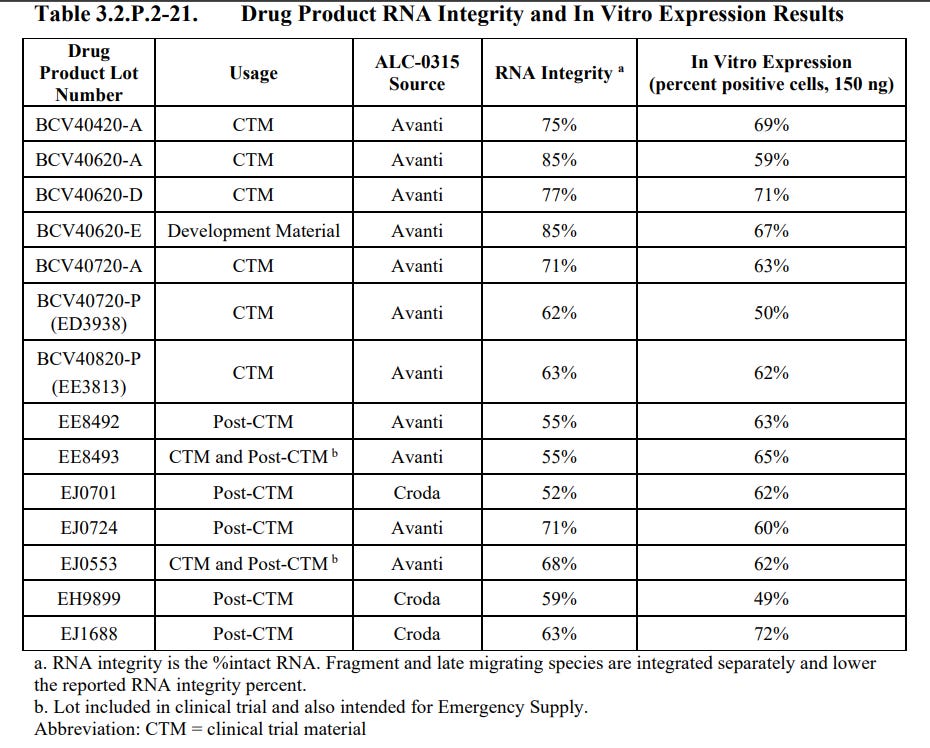
The company declared that “The efficacy of the drug product is dependent on the expression of the delivered RNA, which requires a sufficiently intact RNA molecule.”
“The complete, intact mRNA molecule is essential to its potency as a vaccine,” professor of biopharmaceutics Daan J.A. Crommelin and colleagues wrote in a review article in The Journal of Pharmaceutical Sciences late last year. “Even a minor degradation reaction, anywhere along a mRNA strand, can severely slow or stop proper translation performance of that strand and thus result in the incomplete expression of the target antigen.”
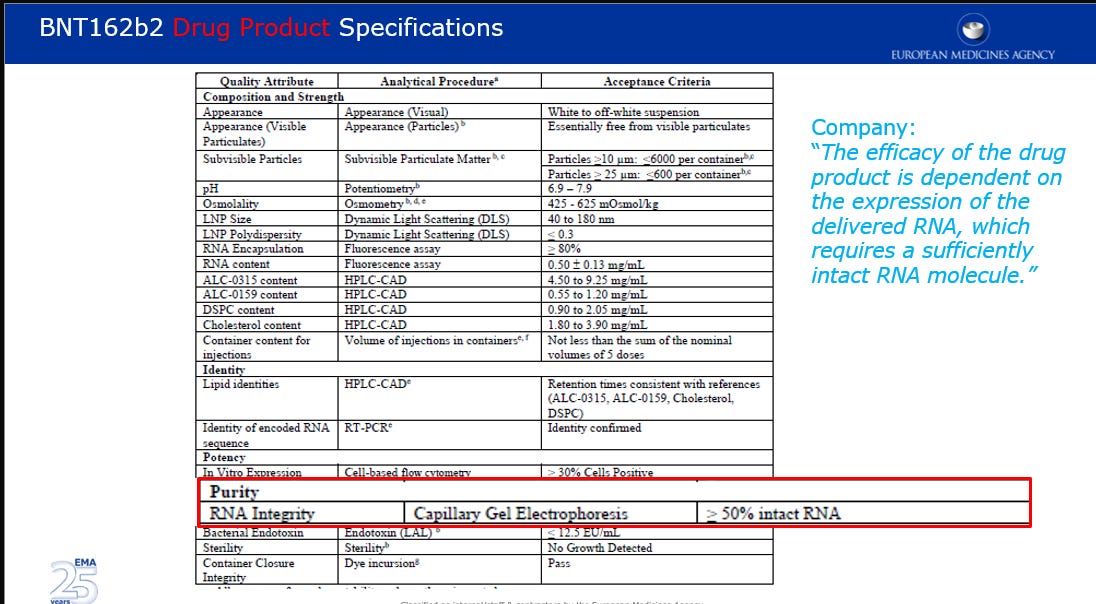
The presence of as much as a 36% variation in batch RNA integrity raised concerns for the European regulators and they flagged this as a Major Objection.

A freedom of information request to the TGA (Australian version of the FDA) just released confirms that they had no idea how to assess an mRNA therapy product. The Australian regulators picked up early signs of degradation in the efficacy too.
Documents relating to the evaluation of the Pfizer COVID-19 vaccine ...
“Antibodies and T cells in monkeys declined quickly after 5 weeks after the second dose of BNT162b2 (V9) raising long term immunity concerns."
and
"There are no distribution and degradation data on the S antigen-encoding mRNA"
https://www.tga.gov.au/sites/default/files/foi-2389-06.pdf
Further to the objection on the RNA integrity, questions were raised in regards to the proposed acceptance criteria of ≥ 30% as a batch release for In-vitro expression and the proposed acceptance criteria of ≥ 80% for RNA encapsulation (in lipid nanoparticles).

SOURCE
COVID-19 mRNA vaccine (nucleoside modified) Rapporteur’s Rolling Review Assessment Report EMA/198107/2020
Degradation During the Process of Manufacture
On 26th November 2020, the EMA met with Pfizer and presented an objection regarding the % of intact mRNA. The document states that the % RNA integrity of the vaccines decreases DURING the process of manufacture, between stages 1, 2 and 3 of the manufacturing process.
Stage 1 : Production of active ingredient : Minimum % of intact RNA is set at 60%
Stage 2 : Addition of adjuvants (additional ingredients) : Minimum % of intact RNA is set at 55%
Stage 3 : Filling of Vials : Finished product is inserted into vials : Minimum % of intact RNA is set at 50%

Effect of % Intact RNA on Toxicity
There is a correlation of 0.56 between % of intact RNA and number of severe reactions per vial. From the EMA documents, we have the % RNA integrity of each batch and the exact size of each batch, and from VAERS we can count the number of severe reactions that each of those batches produced.
"When it is broken, it is less harmful. When it is fully functioning it is lethal."
Post Market Process Modifications Require New Submissions Or Do They …
Major discrepancies in the scale up of the manufacture have been identified including:
% intact mRNA
% In-vitro expression
% encapsulation of mRNA in Lipid Nanoparticles (LNPs)
According to one of the leaked emails dated 25 November, positive news had come from an undisclosed source in the US:
“The latest lots indicate that % intact RNA are back at around 70-75%, which leaves us cautiously optimistic that additional data could address the issue,” the email said.

It’s unclear how the agency’s concerns were satisfied, however ultimately, on 21 December, EMA authorized Pfizer-BioNTech’s vaccine.
The issue was satisfactorily addressed, the agency underlined, when further information was supplied by the manufacturer.
Health Canada told The BMJ that Pfizer had conducted investigations into the root cause of reduced integrity in the commercial vaccine batches, and
“changes were made in their processes to ensure that the integrity was improved and brought in line with what was seen for clinical trial batches.”
Team Enigma, a group of independent researches published on their website howbad.info the following statement:
Some batches/lots are associated with excessive deaths, disabilities and adverse reactions. This variation could be due to -
variation in the amount, meaning the number of doses distributed for a particular lot, OR
variation in the toxicity of the doses
Is it possible that the Pfizer-BioNTech “Process 2” continued to be tweaked until it had reached optimal/very high % of intact mRNA and % expression until they realized that more was more?
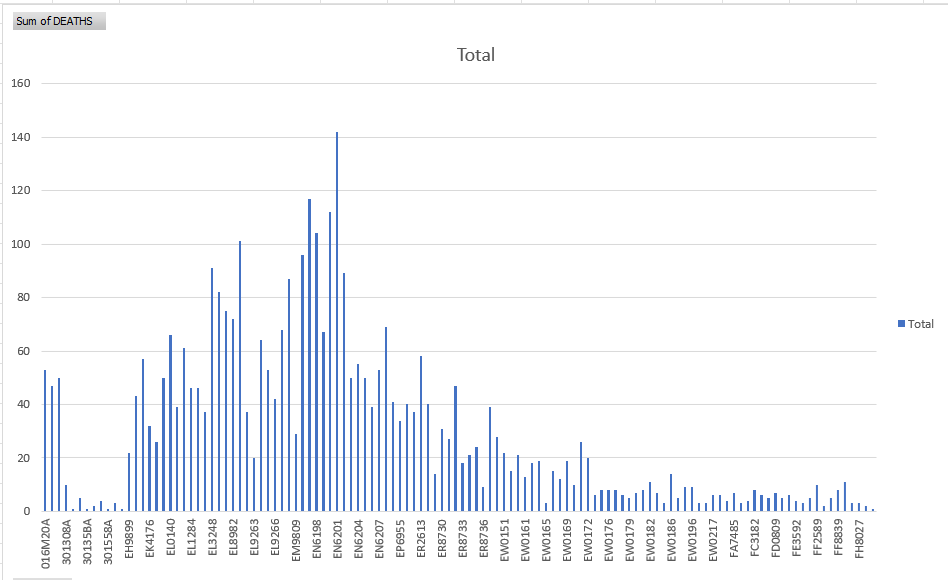
Below is a pivot table of the Pfizer-BioNTech post market batches and their associated sum of deaths taken from VAERs.
We know from the leaked documents that each batch of mRNA equals one batch of product and these batches are in sequential order. Did they keep tweaking the process until a safety signal was triggered?
Leaked documents show that some early commercial batches of Pfizer-BioNTech’s covid-19 vaccine had lower than expected levels of intact mRNA, prompting wider questions about how to assess this novel vaccine platform, writes Serena Tinari.
https://principia-scientific.com/the-ema-data-leak-and-what-it-tells-us-about-mrna-instability/
The BMJ has reviewed the documents, which show that regulators had major concerns over unexpectedly low quantities of intact mRNA in batches of the vaccine developed for commercial production.
If it is not intact mRNA What is the Other 50%?
The EMA—worried about “truncated and modified mRNA species present in the finished product.” Among the many files leaked to The BMJ, an email dated 23 November by a high ranking EMA official outlined a raft of issues.

The Spike protein of SARS-CoV-2 mutations and Real World Effectiveness in the UK for Omicron
It also appears that they knew the efficacy would be affected by the mutations prior to approval and vowed to assess this in real time post launch.
The Spike protein of SARS-CoV-2 undergo mutations, and it thus critically important to investigate the biological significance of these variants in relation to the development of Spike-based covid-19 vaccine candidates. For example, Korber et al. 2020 present evidence that there are now more SARS-CoV-2 viruses circulating in the human population globally that have the G614 form of the Spike protein versus the D614 form that was originally identified from the first human cases in Wuhan, China. Further, Li et al., states that as of May 6, 2020, 329 naturally occurring variants in Spike protein have been reported in the public domain. The applicant is asked to discuss how the chosen Spike antigen variant in BNT162b2 relates to the Spike variants currently on the dominant SARS-CoV-2 viruses circulating in the human population (MS/NOMA OC2). References: Korber et al., 2020: https://www.ncbi.nlm.nih.gov/pmc/articles/PMC7332439/ Li et al., 2020: https://doi.org/10.1016/j.cell.2020.07.012
The UK has 23% of vaccine-free persons. And of Omicron hospitalized, only 26% of persons are vax-free. So we get: about 30-35% vaccine-free persons get about 26% hospitalizations for Omicron.

Further Reading
Moderna Patented CANCER GENE is in Sars-Cov-2 "Spike Protein"
CTCCTCGGCGGGCACGTAG may do just what it was designed to do
Worst Fears Realized: Pfizer mRNA Transcribes into DNA
What the article shows is that in vitro, using a human liver cell line, Pfizer mRNA vaccine uses a natural reverse transcriptase enzyme called LINE-1, and the genetic code of the vaccine is reverse transcribed into the DNA.
Aberrant LINE-1 expression causes cancer and autoimmune disease.
We need experts in this area to step forward and STOP any further use of the 💉until widely studied by independent scientists.


mRNA is stimulating expression of a known cause of these diseases.
LINE-1 and Autoimmune Disease
Hypomethylated and highly expressed LINE-1 has been found in autoimmune diseases such as systemic lupus erythematosus (SLE), Sjögren’s syndrome (SS), and psoriasis (Schulz et al., 2006; Yooyongsatit et al., 2015; Mavragani et al., 2016).
DNA Transcribed from Pfizer mRNA Vaccine Contains MUTANT gp130 Tumor Gene
Spike protein expression is persistant for 60 DAYS (potentially for 15 months according to autopsy)
Histology of mRNA vaccinee lymph nodes shows abundant GCs - Vaccine spike antigen and mRNA persist for weeks in lymph node GCs
*Regulatory Background
European Medicines Agency (EMA) & Centralized marketing authorizations in the European Union
The European Medicines Agency (EMA) is responsible for the scientific evaluation of applications for centralized marketing authorisations in the European Union. This authorisation procedure allows pharmaceutical companies to market the medicine and make it available to patients and healthcare professionals throughout the European Economic Area on the basis of a single marketing authorisation.
To obtain marketing authorisation, medicine developers need to submit specific data on their medicine. EMA then carries out a thorough assessment of these data to decide whether or not the medicine is safe, effective and of good quality and is therefore suitable for use in patients.
A committee of experts, each supported by a team of assessors, evaluates the applications.
In addition to the rapporteur and co-rapporteur, the CHMP also appoints one or more peer reviewers from amongst the CHMP members. Their role is to look at the way the two assessments are performed and ensure that the scientific argumentation is sound, clear and robust.
The CHMP can raise objections or concerns which can relate to any aspect of the medicine. If unresolved, major objections preclude marketing authorisation.
Major objections can relate for example to the way the medicine was studied, the way it is manufactured, or to the effects seen in patients such as the magnitude of the benefits or the seriousness of the side effects.
Nobody found RNA in this. Explain this. https://pseudoscience.substack.com/p/independent-researchers-find-no-mrna